3D Genomic Diversity in Plant Accessions
The first pan-3D genome related to domestication and improvement in plants
During my PhD I worked on a couple of interesting projects related to 3D genomic diversity in plants. In this page I plan to talk about two of them: one which introduced the idea of pan-3D genome in plant accessions and one which performs a detailed investigation between 3D genomic diversity and single nucleotide polymorphisms (SNPs). The following post is currently about the first project, but it will soon be updated with information about the second.
NOTE: The following article only contains part of the results in the pan-3D genome paper.
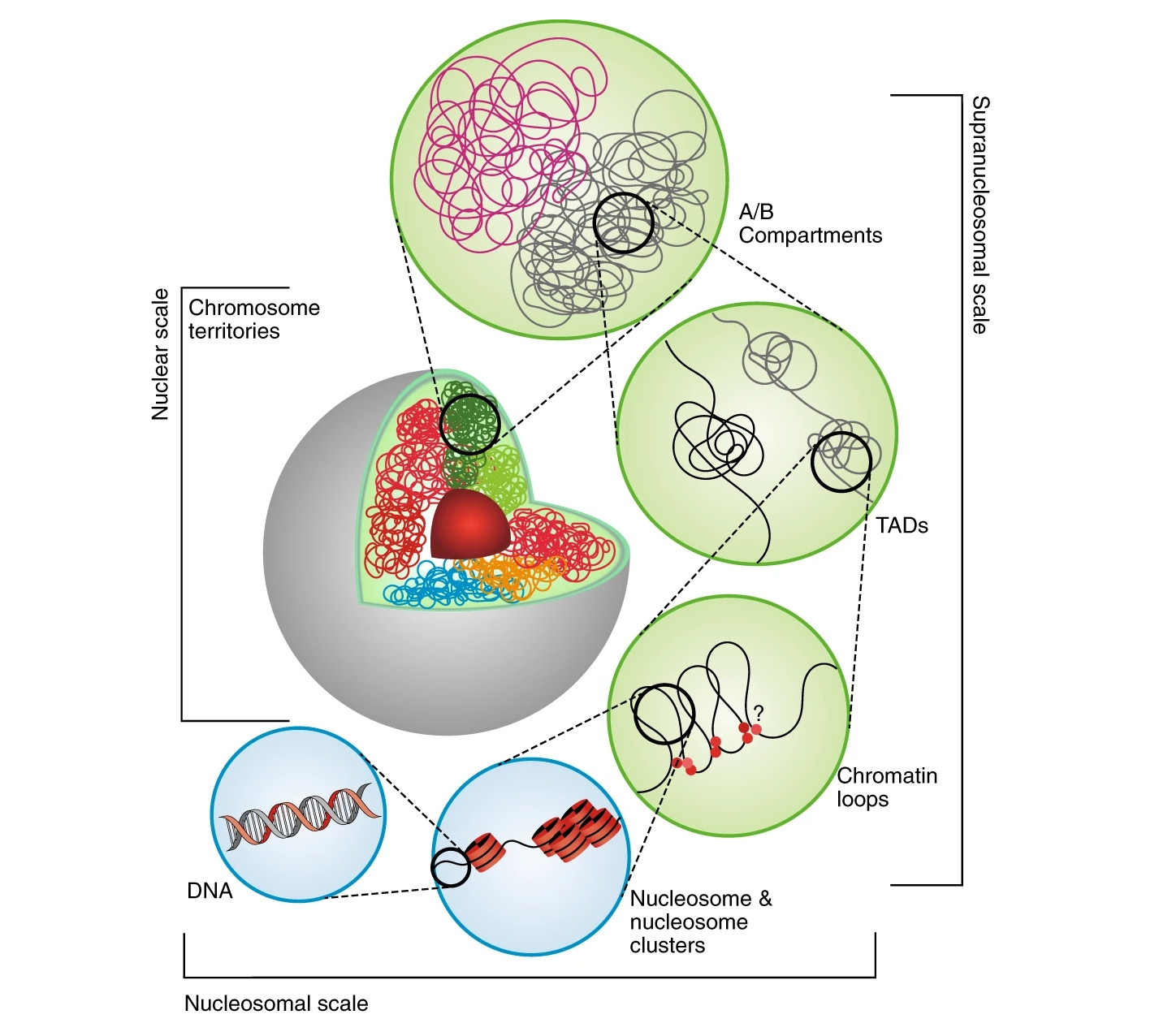
Figure 1: The hierarchical organization of 3D genomes.
3D genome organization can bring distal cis-regulatory elements (CREs) into spatial proximity with their target genes and plays an important role in gene regulation. High-throughput studies have shed light on a hierarchical organization of the 3D genome, which can be arranged into chromosome territories, compartments, topologically associating domains (TADs), and chromatin loops. Recent analyses have suggested that 3D genomes may serve as an important driving force during cell differentiation, embryo development, and genome evolution. However, the diversity of 3D genomes across plant accessions are seldom reported.
Figure 2: Workflow of pan-3D genome analysis.
In this study, we perform the pan-3D genome analysis using Hi-C sequencing data from 27 soybean accessions and comprehensively investigate the relationships between 3D genomic variations and structural variations (SVs) as well as gene expression. This project represents the first studies of 3D genomic diversity investigations across multiple accessions in plant species, and provide a deeper insight into genome evolution and gene regulation in plant accessions.
Data preparation
To investigate the 3D genome organization in soybean, we performed in situ Hi-C sequencing of the 27 accessions which included 3 wild soybeans, 9 landraces, and 15 improved cultivars. The in situ Hi-C experiment for each accession was designed with two biological replicates, and each replicate produced an average of ~470 million raw read pairs and ~100 million unique long-range (more than 20 kb) cis contacts. The two biological replicates of each accession were highly reproducible. Complementing these Hi-C data, we profiled the transcriptome through RNA sequencing (RNA-seq) in these accessions.
3D genome analysis
A/B compartment
Using the in situ Hi-C sequencing data, we checked the organization of A/B compartments and found certain differentiation of A/B compartments within chromosomes. For example, we observed differences around pericentromeric heterochromatin of chromosome 18 in SoyC13 and SoyC03, and variations in the euchromatin arms of chromosome 11 in SoyW02 and SoyC13. Despite these differences, the average A compartment percentages were relatively consistent across individual accessions. Collectively, these results reveal that the overall percentages of compartments are conserved, but the reorganization of A/B compartments within chromosomes occurs frequently in soybean accessions.
Nevertheless, we found that the repeat percentage of the A compartment on several chromosomes was significantly higher than the average level, such as chromosome 1 of SoyC06. Tracing these A compartments showed that the highly repetitive regions mainly came from the regions of intersection between A compartments and B compartments, which were referred to as I regions in this study. We found the A compartments and B compartments from I regions also presented intermediate status. Further investigation using the data from 27 accessions revealed that the GC contents, TSS densities, and repeat percentages of I regions (including the A compartments or B compartments from I regions) exhibited an intermediate status between the A regions and the B regions. Overall, these data illustrate that I regions exhibit intermediate genomic features.
TAD boundary
To identify TADs in soybean, the reported “insulation score” method was adopted in this study. In parallel, contact domains and directionality indexes were also calculated. Although boundaries exhibited some variations across the 27 accessions, they had comparable normalized numbers. The identification results showed that the median size of TADs in soybean was approximately 475 kb.
We analyzed the enrichment of repeats around TAD boundaries. Meta-analysis revealed that repeats were depleted around boundaries. In contrast to humans, LTRs are the dominant TEs in plants. In soybean, non-LTR TEs accounted for less than 5% of the TEs, while LTRs accounted for ~80%. Subsequently, we investigated the enrichment of different repeat types. Interestingly, although most repeats were found to be depleted around boundaries, LINEs and SINEs were found to be enriched around boundaries particularly. These results were further confirmed through the analyses of each of the 27 soybean accessions. It has been found that most identified LINEs in plants come from the L1 and RTE superfamilies. Enrichment analysis of superfamilies showed that the length of the L1 fragment was longer around boundaries.
Pan-3D genome analysis
A/B compartment
We performed pan-analyses of A/B compartments through the investigation of their conservation and variation across the 27 soybean accessions. It showed that 78.5% of the compartment bins in the genome exhibited conservation. Modeling the pan-3D genome size by iteratively randomly sampling accessions suggested a closed pan-3D genome with finite numbers of both conservative and variable compartments, which showed a similar pattern to our previous pan-genome analyses. The A compartments showed a higher ratio of conservative compartments than B compartments. We then inspected the variable compartments by dividing them into three types: compartments with only A compartments, compartments with only B compartments, and compartments with both A and B compartments (AB variable compartments). A large proportion (64.83%) of the compartment variation occurred in AB variable compartments, indicating extensive compartment switching across soybean accessions.
TAD boundary
We performed the pan-3D genome analysis of boundaries by adopting a method combining the alignment and clustering of boundaries across 27 accessions. We found that nearly 20% (708/4,505) of the boundary clusters were categorized as core types, 41.82% (1884/4505) were categorized as dispensable types, and 42.46% (1913/4505) were categorized as private types. GO analysis showed that the core types were mainly involved in basic metabolism and transcription processes, while the dispensable types were involved in various noncoding RNAs and posttranscriptional RNA processing. No significant enrichment was found for the private types, indicating that private boundaries may participate in specific biological functions.
Genomic feature analyses demonstrated that the common/core boundaries had the highest TSS density and lowest GC content and repeat percentage. We then investigated the enrichment of different repeat types around boundaries of the pan-3D genome. Surprisingly, two opposite patterns were observed. The first pattern showed a higher proportion of repeat elements in the specific boundaries than in the core and dispensable boundaries, such as Gypsy elements and satellite repeats, indicating that these two types of elements may play an important roles in specific boundary formation. In contrast, the second pattern showed a higher proportion of repeats in the core boundaries, including LINEs, SINEs, and all DNA transposons except Helitron elements. Additionally, the distributions of several repeat types, such as Copia elements and Helitron elements, seemed to be unclear. Moreover, we found that LINEs and SINEs were enriched around the boundaries of core and dispensable types but showed a depleted pattern around specific boundaries.
Structural variation analysis
We next investigated the contributions of these different types of SVs to 3D genome variation. Each type of SVs contributed to boundary variations significantly. Nevertheless, less than half of the boundary variations could be independently explained by SVs, among which PBA-PAV showed the largest (42.05%) contribution. Additionally, ABA-PAV contributed 4.97% of the boundary variations, which was second only to the contribution of PBA-PAV. These results indicated that PAVs, not CNVs, INVs, or TRANSs, played the most important roles in boundary variations. Next, we checked the effect of SVs on boundary variations. As expected, ABA-PAV showed the largest effect on boundary variations. Interestingly, for most balanced rearrangements, such as INVs or TRANSs, we did not observe extensive conservation of boundaries with either ABA types or PBA types, implying that boundaries were not solely dependent on sequence conservation during genomic rearrangement across soybean accessions. Together, these results reveal that at the global level, there are significant correlations between SVs and 3D genome variations but relatively finite changes of 3D genomes, indicating that SVs play a contributory but not deterministic role in 3D genome variations.
TE insertion is an important contributor driving SV formation. Hence, we investigated the effects of TEs in driving SVs causing 3D genome variation. For most TEs, we did observe a lower percentage in ABA or PBA SVs than in NBA SVs. Similar patterns were also observed for most TRs. Only Gypsy elements and satellite repeats showed extremely significant enrichment in ABA SVs relative to PBA SVs, consistent with the results showing that these two repeats were enriched in private boundaries.
Selection analysis
Using the pan-3D genome data, we observed a core set of over 1800 TAD boundary clusters shared by all three soybean groups. In addition, 457, 521, and 1091 specific TAD boundary clusters were identified for individual G. soja, landrace, and improved cultivar groups. Combining with pairwise population differentiation level (FST) across different soybean groups, we found the selection of TAD boundaries was higher than that of non-boundaries during the soybean domestication, whereas no significant difference during improvement. Further investigation suggested that the genes around selected TAD boundaries had higher expression levels than those of unselected TAD boundaries.
We found the expression variations of some domesticated genes were closely related to the 3D genomic variations. For example, SoyZH13_14G139200 was located in a domesticated selection sweep region. Structural variations led to the loss of the TAD boundary in two wild soybeans SoyW02, SoyW03, and one landrace SoyL02. As a result, we observed that the SoyZH13_14G139200 showed significant higher expression level in these 3 accessions than the others. These data suggested that 3D genome structure variation may also play an important role in soybean domestication and improvement.
Conclusions
- The first pan-3D genome related to plant domestication and improvement indicates conservative A/B compartments and dynamic TAD boundaries across plant accessions.
- I regions largely contribute to A/B compartment divergence.
- Non-LTR retrotransposons maintain TAD boundaries, and Gypsy elements and satellite repeats establish private TAD boundaries.
- PAV is the major contributor to 3D genome variations.
- 3D genome may undergo selection during domestication and improvement.
Where should you look to learn more?
More details can be found in our pan-3D genome paper. The sequencing data generated in this study are available at the Genome Sequence Archive (GSA, https://bigd.big.ac.cn/gsa/; accession number: PRJCA009364). Computational scripts used for data analyses conducted as part of this study are available under MIT license at Github: https://github.com/LingbinNi/soybean_pan-3D_genome_analysis and Zenodo: https://doi.org/10.5281/zenodo.7514511. Custom UCSC chain files between ZH13 and 26 query genomes: https://figshare.com/articles/dataset/UCSC_chain_files_of_soybean_genomes/20027336.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFF1000101-3), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA24030501), and the National Natural Science Foundation of China (31788103, U22A20473).
References
Ni L, Liu Y, Ma X, Liu T, Yang X, Wang Z, Liang Q, Liu S, Zhang M, Wang Z, et al. (2023). Pan-3D genome analysis reveals structural and functional differentiation of soybean genomes. Genome Biol 24, 12.